Molecular Simulation Research
Molecular Simulations

Faculty: Thomas A. Knotts
Controlling the behavior of physical systems is at the core of chemical engineering. The behavior observed on the macroscopic scale is the aggregate of countless interactions that occur at the molecular level. An understanding of what occurs on these small length scales enables prediction, manipulation, and control of the behavior of larger systems. Molecular modeling and simulation consist of a suite of computational techniques that can be used to create individual atoms and molecules in a virtual environment and "watch" what happens. With the resources available at the Fulton Supercomputing Laboratory at BYU, molecular simulation and modeling tools are used to investigate many systems of interest including the phase behavior of fluids, the dynamics of ion movement in fuel cells, the interactions of proteins on surfaces, and the prediction of thermophysical properties of industrially-relevant compounds. Additionally, significant effort is devoted to development of novel tools, algorithms, and theories. Such are required as the problems being investigated are complex and unique. Please visit the links below for more details.
More information:
Multistate Folding of Proteins
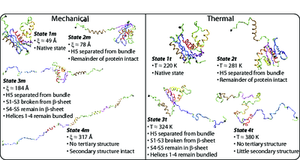
Faculty: Thomas A. Knotts
Proteins are involved in almost all aspects of life. A protein begins as a long linear chain of amino acids but becomes a compact, three-dimensional structure in a process called "folding." Understanding the folding process is one of the most important topics in modern biochemistry. Protein structure can be destroyed (a process called "denaturation" or "unfolding") using external influences such as pH, temperature, and pressure. One important question in regards to folding/ unfolding is whether different external influences change the folding mechanisms.
More information:
Two-phase Molecular Dynamics Simulation

Molecular dynamics simulations are being performed on long-chain alcohols using intermolecular potentials that have been developed within our research group from ab initio calculations. The simulations are performed at conditions below the critical point where two phases coexist. The simulation is performed at several different temperatures to identify the equilibrium densities of the coexisting liquid and vapor at several temperatures close to the critical temperature.